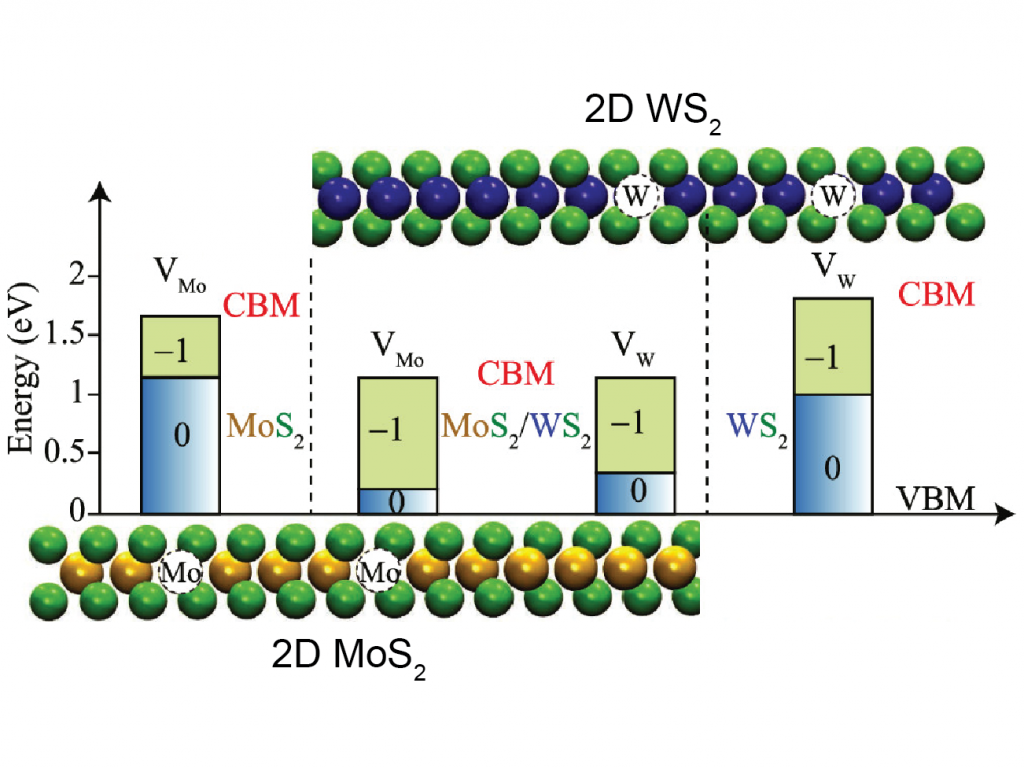
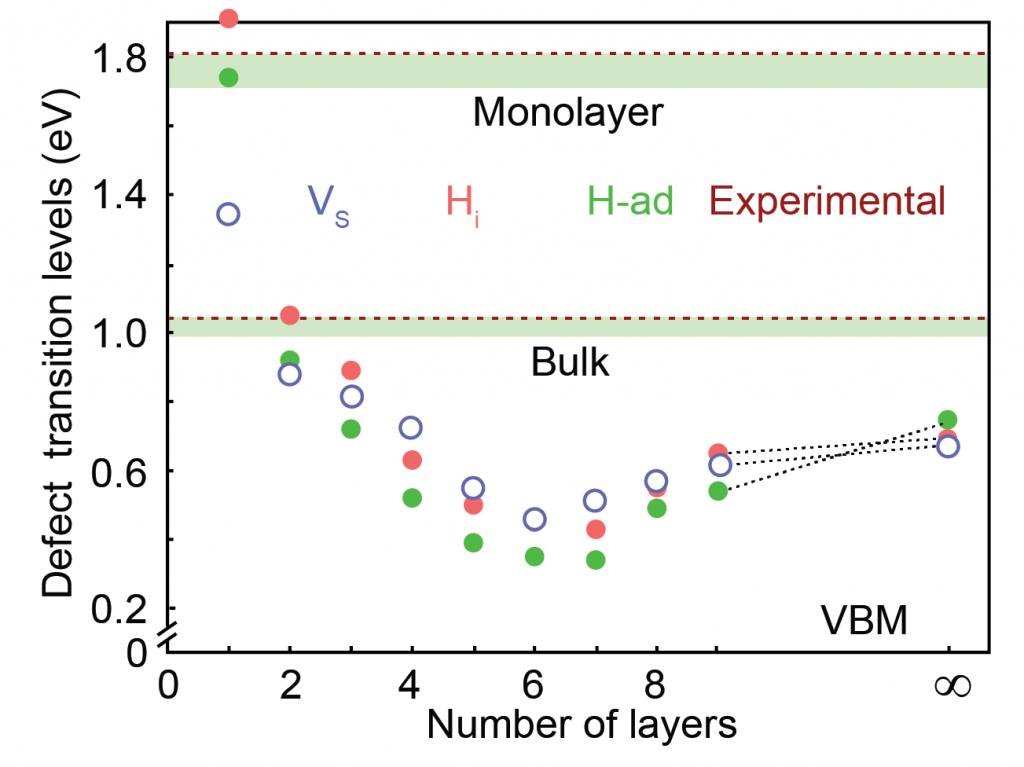
Defects in solid state have a significant impact on the thermodynamic, electronic, optical, and magnetic properties of the material. Using first principles methodology, we investigate the thermodynamic properties of doping and defects in semiconductors. For many years, scientists debated the origin of observed n-type conductivity in MoS2. Although, n-type conductivity was previously attributed to sulphur vacancy, our study demonstrates the hydrogen impurity behaving as shallow donors, and hence resulting in n-type conductivi [1]. Similarly, the extrinsic and intrinsic defects in 2D-WS2 has been studied in hybrid functional level theory. Sulphur vacancy (VS) is identified as an electron trap in contradiction to previous notion of it being a shallow donor and hydrogen interstitial is found to be shallow donor[2].
Accurate description of solubility and defect ionization energies in low dimensional nanostructures is critical for electronic applications of semiconductors with improved functionalities. Changing the number of layers [3], hetero-structuring without changing the integrity of the 2D materials [4], varying the diameter of 1D nanowires [5] are demonstrated as effective strategies for tuning thermodynamic and electronic properties arising from defects and dopants.
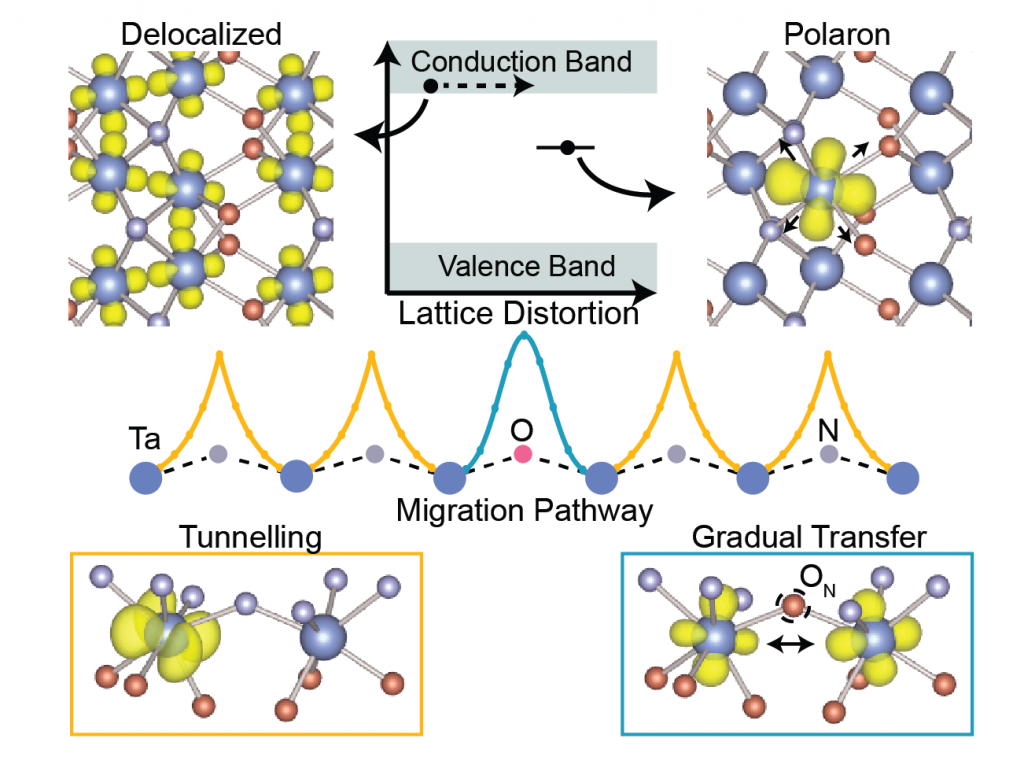
Moreover, electronic perturbation can also act as doping without the insertion of any foreign atoms in the lattice. The perturbation potential around any atom helps to capture electron to behave as “self-trapped” small polaron. We found the formation of small electron polaron in tantalum oxynitride (β-TaON). The trade-off between adiabatic and non-adiabatic hopping migration of small polarons supports the experimentally observed low mobility and high carrier lifetime in the β-TaON photoanode. [6]
Engineering Defect Transition-Levels through van der Waals Heterostructure J. Phys. Chem. C 122, 24475, (2018)
Origin of layer-dependent electrical conductivity of transition metal dichalcogenides, Phys. Rev. B, 105, 165430 (2022)
Formation of Small Electron Polaron in Tantalum Oxynitride: Origin of Low Mobility, J. Phys. Chem. C, 125, 11548-11554 (2021)
3. A. Singh, M. Dey, and A. K. Singh, Origin of layer-dependent electrical conductivity of transition metal dichalcogenides, Phys. Rev. B, 105, 165430 (2022)
4. A. Singh, A. Manjanath and A. K. Singh, Engineering Defect Transition-Levels through van der Waals Heterostructure, J. Phys. Chem. C 122, 24475, (2018)
5. M. Dey, S. Chowdhury, S. Kumar, and A. K. Singh. Quantum confinement effect on defect level of hydrogen doped rutile VO2 nanowires. J. Appl. Phys. 131, 235702 (2022)